

Μειώνει η χαμηλή «κακή» χοληστερίνη τον καρδιαγγειακό κίνδυνο;
8 Αυγούστου 20
Θετικό δείγμα covid-19 σε αιμοκαθαιρόμενη ασθενή στο νοσοκομείο Μυτιλήνης.
18 Αυγούστου 20ΚΛΗΡΟΝΟΜΙΚΕΣ ΝΕΦΡΟΠΑΘΕΙΕΣ: Από το παιδί στον ενήλικα

Οι κληρονομικές νεφροπάθειες περιλαμβάνουν μία ολόκληρη σειρά κληρονομικής φύσεως γενετικών νοσημάτων των νεφρών και παρ’ όλες τις εντυπωσιακές επιτεύξεις της γενετικής και βιολογίας, αρκετά παραμένουν αδιευκρίνιστα ως προς την αιτιολογία και παθογένεια, κυρίως όμως παραμένουν δυσδιάγνωστα και άνευ θεραπείας. Από την μεγάλη ομάδα των κληρονομικών νεφροπαθειών, οι πλέον συχνές που διαγιγνώσκονται συνήθως στην παιδική ηλικία και αντιμετωπίζονται κυρίως στην ενήλικο ζωή είναι από τις διαταραχές του κολλαγόνου της βασικής μεμβράνης των νεφρικών σπειραμάτων, το σύνδρομο ALPORT και η νόσος λεπτής βασικής μεμβράνης και από τις κληρονομικές κυστικές παθήσεις, η πολυκυστική νεφρική νόσος.
Α. Μητσιώνη, Παιδίατρος -Παιδονεφρολόγος, Διευθυντής ΕΣΥ, Νεφρολογικό Τμήμα, Νοσοκομείο Παίδων Παναγιώτη & Αγλαΐα Κυριακού
ΔΙΑΤΑΡΑΧΕΣ ΤΟΥ ΚΟΛΛΑΓΟΝΟΥ IV ΤΗΣ ΒΑΣΙΚΗΣ ΜΕΜΒΡΑΝΗΣ
Το COL IV είναι το κύριο συστατικό των βασικών μεμβρανών και αποτελείται από 6 αλύσους που ονομάζονται α1, α2, α3, α4, α5, α6 (IV) που κωδικοποιούνται από τα αντίστοιχα γονίδια COL IV A1 – COL IV A6 και τα 6 γονίδια των αλύσων αυτών μοιράζονται ανά ζεύγη σε 3 χρωμοσώματα. Οι άλυσοι α1 και α2 κωδικοποιούνται από τα γονίδια COLVI A1 και A2 εδράζονται στο χρωμόσωμα 13 q, οι άλυσοι α3 και α4 από τα γονίδια COL IVA4 και A5 στο χρωμόσωμα 2q και οι άλυσοι α5 και α6 από τα γονίδια COL IV A5 και A6 στο Xq χρωμόσωμα. Μεταλλάξεις των γονιδίων αυτών ανιχνεύονται στο σύνδρομο ALPORT και στη νόσο της λεπτής βασικής μεμβράνης.
Σύνδρομο ALPORT
Το σύνδρομο ALPORT είναι κληρονομική νεφρίτιδα με φαινοτυπική και γενετική ετερογένεια. Η συχνότητα του γονιδίου είναι 1:5000 – 1:10000, αποτελεί το 3% των αιτίων ΤΣΝΑ. Έχουν περιγραφεί τρείς τύποι κληρονομικότητας του συνδρόμου. Στο 80-85% των περιπτώσεων είναι Χ-φυλοσύνδετος χαρακτήρας και σχετίζεται με μεταλλάξεις των α5 και α6 αλυσίδων του COL ΙV. Στο 5-10% είναι αυτοσωματικός υπολειπόμενος χαρακτήρας και στο 3-5% των περιπτώσεων είναι αυτοσωματικός επικρατούν χαρακτήρας και σχετίζονται με μεταλλάξεις των α3 και α4 αλυσίδων του COL IV. Σε 10% των πασχόντων η νόσος οφείλεται σε de novo μεταλλάξεις. Στο φυλοσύνδετο τύπο, οι κλινικές εκδηλώσεις είναι πλέον τυπικές, αφορούν κυρίως τους άνδρες που πάσχουν και καταλήγουν σε ΧΝΑ. Στις γυναίκες η εξέλιξη της νόσου ποικίλλει, συνήθως είναι ασυμπτωματικοί φορείς με ήπια μικροσκοπική αιματουρία αλλά είναι δυνατόν να εξελιχθεί σε νεφρική ανεπάρκεια σε μεγάλη ηλικία. Στον αυτοσωματικό τύπο κληρονομικότητας δεν παρατηρείται διαφορά μεταξύ ανδρών και γυναικών ως προς την κλινική εικόνα και την εξέλιξη της νόσου. Στον υπολειπόμενο τύπο για την εμφάνιση της νόσου απαιτούνται δύο κληρονομούμενες μεταλλάξεις, μία από κάθε γονέα, ενώ ο επικρατούν τύπος φαίνεται ότι οφείλεται σε μία βαρύτερη μετάλλαξη.

Οι κύριες κλινικές εκδηλώσεις της νόσου είναι:
Aιματουρία: είναι σταθερό σύμπτωμα της νόσου, είναι επιμένουσα μικροσκοπική με ή χωρίς επεισόδια μακροσκοπικής αιματουρίας. Στο φυλοσύνδετο τύπο εμφανίζεται σε όλους τους άνδρες σε νεαρή ηλικία ενώ στις γυναίκες εκδηλώνεται αργότερα και σε ποσοστό 90%. Σημειώνεται ότι 10% των γυναικών φορέων της νόσου δυνατόν να μην παρουσιάζουν αιματουρία. Στον αυτοσωματικό τύπο, υπολειπόμενο και επικρατούντα, εμφανίζεται με την ίδια συχνότητα και στα δύο φύλα.
Λευκωματουρία: δεν είναι απαραίτητο στοιχείο του συνδρόμου, η εμφάνιση της αυξάνει με την ηλικία, στο φυλοσύνδετο τύπο παρατηρείται στους άνδρες ενώ στον υπολειπόμενο και στα δύο φύλα. Σε 30% των περιπτώσεων δυνατόν να εκδηλωθεί με νεφρωσικό σύνδρομο. Λευκωματουρία χωρίς αιματουρία δεν έχει περιγραφεί.
Διαταραχές ακοής: η βαρηκοΐα είναι αμφοτερόπλευρη, εξελικτική, νευροαιαισθητήρια, επηρεάζει τις μέσες και υψηλές συχνότητες πέρα των 2000-8000Hz, αφορά και τα δύο φύλα και συνοδεύει τις βαρύτερες περιπτώσεις. Δεν είναι συγγενής, συνήθως εμφανίζεται στο τέλος της παιδικής ηλικίας. Η βλάβη εδράζεται στο κολλαγόνο του κοχλία.
Οφθαλμολογικές διαταραχές: αφορούν το φακό, τον αμφιβληστροειδή και τον κερατοειδή. Εμφανίζονται συνήθως μετά την παιδική ηλικία και δεν έχουν παρατηρηθεί σε ασθενείς χωρίς βαρηκοΐα. Ο πρόσθιος φακόκωνος, όταν παρουσιάζεται, θεωρείται παθογνωμονικός. Είναι αμφοτερόπλευρος, πρόκειται για προβολή του φακού στον πρόσθιο θάλαμο και σύμφωνα με μελέτες, οφείλεται στην απουσία των α3, α4 και α5 αλύσων του κολλαγόνου IV στην πρόσθια κάψα του φακού. Οι ωχρικές και περιωχρικές κηλίδες του αμφιβληστροειδούς είναι χαρακτηριστικές και δεν προκαλούν διαταραχές στην όραση. Δυνατόν επίσης να εμφανιστεί καταρράκτης και οπίσθια πολύμορφη δυστροφία του κερατοειδούς
Νεφρική ανεπάρκεια: παρατηρείται σε 100% των ανδρών σε οποιοδήποτε τύπο κληρονομικότητας, ενώ στις γυναίκες το ποσοστό εξαρτάται από την κληρονομικότητα. Έχουν την ίδια κατάληξη στον αυτοσωματικό τύπο, επικρατούντα και υπολειπόμενο, ενώ στο φυλοσύνδετο μικρό ποσοστό φθάνει στο τελικό στάδιο σε προχωρημένη ηλικία. Εάν εμφανιστεί ΤΣΝΑ πριν την ηλικία των 30 ετών χαρακτηρίζεται ως σύνδρομο ALPORT νεανικού τύπου, εάν εμφανιστεί σε ηλικία >30 ετών ενήλικου τύπου.
Δυνατόν οι πάσχοντες από σύνδρομο ALPORT να παρουσιάσουν λειομυομάτωση του οισοφάγου και άλλων οργάνων και αιματολογικές διαταραχές όπως μεγαθρομβοκυτταρο-πενία. Η διάγνωση του συνδρόμου βασίζεται στην κλινική εικόνα (νεφρικές και εξωνεφρικές εκδηλώσεις), στο οικογενειακό ιστορικό, στην εξέταση του νεφρικού ιστού με ηλεκτρονικό μικροσκόπιο (ΗΜ), στη ανισοϊστοχημική μελέτη του νεφρικού ιστού και της επιδερμίδας και στη γονιδιακή ανάλυση που σπανίως απαιτείται και είναι δυσχερής. Η ιστολογική εξέταση του νεφρικού ιστού μετά από νεφρική βιοψία πρέπει να γίνεται με ΗΜ δεδομένου ότι οι βλάβες που παρατηρούνται στο οπτικό μικροσκόπιο δεν είναι παθογνωμονικές. Οι χαρακτηριστικές βλάβες στο ΗΜ είναι ακανόνιστη πάχυνση της βασικής μεμβράνης με παχύνσεις, λεπτύνσεις και διαχωρισμό σε στρώματα (splitting – lamellation) προσδίδοντας πεταλιώδη εμφάνιση. Στην ανισοϊστοχημική μελέτη, με τη βοήθεια μονοκλωνικών αντισωμάτων κατά των α3, a4, a5 COL(IV) αλύσων και τη μέθοδο του έμμεσου ανοσοφθορισμού, ανιχνεύεται και αναγνωρίζεται η κατανομή των αλύσεων αυτών στις βασικές μεμβράνες των διαφόρων τμημάτων του νεφρώνα και στη βασική μεμβράνη της επιδερμίδας.
Δεδομένου δε, ότι φυσιολογικά η κατανομή των 6 αλύσων του κολλαγόνου ΙV είναι διαφορετική στα διάφορα τμήματα του νεφρώνα και ότι στη βασική μεμβράνη της επιδερμίδας υπάρχει α5 αλυσίδα φυσιολογικά, με την ανισοϊστοχημική μέθοδο είναι δυνατή όχι μόνο η διάγνωση της νόσου αλλά επιπλέον και η διάκριση μεταξύ του φυλοσύνδετου και του αυτοσωματικού τύπου της κληρονομικότητας. Το σύνδρομο ALPORT πρέπει να διαφοροδιαγιγνώσκεται από τη νόσο της λεπτής βασικής μεμβράνης, την IgA νεφροπάθεια, την οξεία μεταλοιμώδη σπειραματονεφρίτιδα και την αιματουρία από αποφρακτική νόσο, ουρολοίμωξη ή ασβεστιουρία.
Ειδική θεραπεία της νόσου δεν υπάρχει.
Εάν υπάρχει λευκωματουρία η χορήγηση αναστολέων/ ανταγωνιστών του μετατρεπτικού ενζύμου φαίνεται ότι επιφέρει μία σχετική καθυστέρηση στην εξέλιξη της νόσου. Οι νεώτερες οδηγίες της Αμερικανικής Παιδιατρικής Νεφρολογίας συνιστούν την έναρξη της θεραπείας και με παθολογικές τιμές μικρολευκωματίνης ούρων, εάν προκύπτει από το οικογενειακό ιστορικό σύνδρομο ALPORT νεανικού τύπου. Υπάρχουν επίσης αρκετές μελέτες όπου έχει χορηγηθεί κυκλοσπορίνη, τα αποτελέσματα όμως είναι αντικρουόμενα. Οι πάσχοντες από σύνδρομο ALPORΤ όταν καταλήξουν σε ΤΣΝΑ δύνανται να μεταμοσχευτούν. Οι εξωνεφρικές εκδηλώσεις δεν υποχωρούν μετά τη νεφρική μεταμόσχευση. Όσον αφορά την μεταμόσχευση από ζώντα δότη, πρέπει να αποκλείονται οι φορείς της νόσου (όταν η νόσος μεταβιβάζεται με τον φυλοσύνδετο χαρακτήρα) δεδομένου ότι αναφέρονται στη βιβλιογραφία δότες που εμφάνισαν ΧΝΑ μετά τη δωρεά του οργάνου. Τονίζεται ότι 10% των φορέων γυναικών του φυλοσύνδετου τύπου δυνατόν να μην παρουσιάζουν αιματουρία. Μετά τη νεφρική μεταμόσχευση μπορεί να παρουσιαστεί υποτροπή της νόσου στο μόσχευμα λόγω ανάπτυξης αντισωμάτων κατά της βασικής μεμβράνης των σπειραμάτων. Είναι επιπλοκή που παρατηρείται τους πρώτους μήνες έως τον πρώτο χρόνο μετά τη μεταμόσχευση, σε ποσοστό 3-5% των μεταμοσχεύσεων, υπάρχει κίνδυνος εξέλιξης προς ταχέως εξελισσόμενη σπειραματονεφρίτιδα και χαρακτηρίζεται ιστολογικά από γραμμοειδείς εναποθέσεις IgG κατά μήκος της βασικής μεμβράνης των σπειραμάτων. Δυνατόν να συνοδεύεται από πνευμονική αιμορραγία (σύνδρομο Goodpasture). H πρόγνωση είναι επιφυλακτική.
Nόσος της λεπτής βασικής μεμβράνης

Η συχνότητα της νόσου είναι δύσκολο να καθοριστεί, υπολογίζεται περίπου σε 1% του πληθυσμού, δυνατόν όμως να υποεκτιμάται. Κληρονομείται με επικρατούντα σωματικό χαρακτήρα, ενώ το 1/3 των ασθενών έχουν de novo μετάλλαξη. Σχετίζεται με μεταλλάξεις των COL IV A3 και COL IV A4 γονιδίων. Οι πάσχοντες είναι ετερόζυγοι φορείς της μετάλλαξης κληρονομούμενης από τον ένα γονέα. Οι ετερόζυγες μεταλλάξεις σε ασθενείς με νόσο λεπτής βασικής μεμβράνης είναι παρόμοιες ή ίδιες με αυτές που παρατηρούνται στο σύνδρομο ALPORT που κληρονομείται με τον υπολειπόμενο χαρακτήρα.
Έχει παρατηρηθεί όμως οι ίδιες μεταλλάξεις να προκαλούν διαφορετική κλινική εικόνα σε μέλη της ίδιας οικογένειας. Επομένως ασθενείς με μετάλλαξη του COL IV A3 ή COL IV A4 γονιδίου του χρωμοσώματος 2 δυνατόν να είναι ετεροζυγώτες με νόσο λεπτής βασικής μεμβράνης ή φορείς αυτοσωματικού υπολειπόμενου συνδρόμου ALPORT. Αυτοσωματικό με επικρατούντα χαρακτήρα ALPORT επίσης οφείλεται σε μετάλλαξη των COL IV A3/A4 γονιδίων αλλά δεν είναι γνωστό γιατί παρατηρείται αυτή η μεγάλη διαφορά στη κλινική έκφραση. Τα γονίδια αυτά είναι τεράστια σε μέγεθος και έχουν ταυτοποιηθεί μεταλλάξεις σε 12-17% των ατόμων με νόσο της λεπτής βασικής μεμβράνης ενώ το ποσοστό αυξάνει σε 67% όταν αφορά οικογένειες όπου είναι ήδη γνωστή η υπεύθυνη μετάλλαξη.
Κλινικά χαρακτηρίζεται από επίμονη μικροσκοπική αιματουρία. Επεισόδια μακροσκοπικής αιματουρίας παρατηρούνται σε ποσοστό 5-22%των ασθενών. Η ηλικία διάγνωσης ποικίλλει από τον πρώτο χρόνο ζωής μέχρι πολύ μεγάλη ηλικία. Δυνατόν να παρατηρηθεί μικρού βαθμού λευκωματουρία έως 500 mg σε ενήλικες. Αρτηριακή υπέρταση παρουσιάζουν τα 11-30% των ενηλίκων και η νεφρική λειτουργία είναι φυσιολογική. Βασικά πρέπει να γίνει διαφορική διάγνωση από την IgΑ νεφροπάθεια και το σύνδρομο ALPORT. Bασίζεται στο οικογενειακό ιστορικό, στην ύπαρξη ή όχι εξωνεφρικών εκδηλώσεων (που όμως εμφανίζονται αργότερα στην εφηβεία) και στα ευρήματα από τη βιοψία νεφρού στην εξέταση με ηλεκτρονικό μικροσκόπιο. Ιστολογικά, σύμφωνα με την WHO, η νόσος χαρακτηρίζεται από ομοιόμορφα λεπτή βασική μεμβράνη, πάχους <250 mm σε ενήλικες και <180 mm σε παιδιά 2-11 ετών σε ποσοστό > 50% των σπειραμάτων.
Η διάγνωση συχνά είναι δυσχερής και σημειώνεται ότι και στα αρχικά στάδια του συνδρόμου ALPORT δυνατόν η βασική μεμβράνη των σπειραμάτων να είναι λεπτή. Στην ανοσοϊστοχημεία νεφρικού ιστού και επιδερμίδας παρατηρούνται σε ποσοστό 20-30% ψευδώς αρνητικά αποτελέσματα. Η εμφάνιση της νόσου λεπτής βασικής μεμβράνης προϋποθέτει την ύπαρξη ετερόζυγης μετάλλαξης ενώ οι ομοζυγώτες ή διπλοί ετεροζυγώτες των μεταλλάξεων παρουσιάζουν σύνδρομο ALPORT υπολειπόμενου χαρακτήρα. Εξηγείται σαν dose – effect φαινόμενο, όπου η απουσία ενός φυσιολογικού αλληλόμορφου γονίδιου οδηγεί σε μικρότερη παραγωγή του α3α4α5 τριμερούς, ενώ η απουσία και των δύο αλληλομόρφων γονιδίων οδηγεί σε πλήρη έλλειψη του α3α4α5 τριμερούς και την εμφάνιση του σ. ALPORT. Πιθανόν το με επικρατούντα χαρακτήρα σ. ALPORT να οφείλεται σε βαρύτερη μετάλλαξη που δυνατόν να εκδηλώνεται και στην ετερόζυγο κατάσταση. Οι ασθενείς με νόσο λεπτής βασικής μεμβράνης πρέπει να παρακολουθούνται συστηματικά για πιθανότητα ανάπτυξης αρτηριακής υπέρτασης.
ΠΟΛΥΚΥΣΤΙΚΗ ΝΕΦΡΙΚΗ ΝΟΣΟΣ
Αν και νεφρικές κύστεις παρατηρούνται σε πολλά νεφρικά νοσήματα, ο όρος πολυκυστική νεφρική νόσο αφορά:
1) την πολυκυστική νεφρική νόσο επικρατούντα χαρακτήρα (ΕΠΚΝ) που κληρονομείται με τον επικρατούντα αυτοσωματικό χαρακτήρα και χαρακτηρίζεται από σχηματισμό κύστεων σε οποιοδήποτε τμήμα των νεφρώνων, κύστεων που δεν επικοινωνούν μεταξύ τους και δυνατόν να συνοδεύεται από εξωνεφρικές εκδηλώσεις όπως παρουσία κύστεων στο ήπαρ και πάγκρεας και ενδοκρανιακά ανευρύσματα και
2) την πολυκυστική νεφρική νόσο υπολειπόμενου χαρακτήρα (ΥΠΚΝ) που κληρονομείται με τον αυτοσωματικό υπολειπόμενο χαρακτήρα και χαρακτηρίζεται από σχηματισμό κύστεων στο αθροιστικά σωληνάρια, κύστεων που επικοινωνούν μεταξύ τους και συνυπάρχει συγγενής ηπατική ίνωση. Η ΕΠΚΝ έχει συχνότητα 1:400 -1000 γεννήσεις, διαγιγνώσκεται συχνότερα στον ενήλικα αλλά δυνατόν να εμφανιστεί και στην παιδική ηλικία και ενδομήτρια ζωή. Προκαλείται σε ποσοστό 85%, από μετάλλαξη του PKD1 γονιδίου που εδράζεται στο χρωμόσωμα 16 και κωδικοποιεί την πολυκυστίνη 1 και σε ποσοστό 15% από μετάλλαξη του PKD2 γονιδίου που εδράζεται στο χρωμόσωμα 4 και κωδικοποιεί την πολυκυστίνη 2. Οι ασθενείς με μεταλλάξεις στο γονίδιο PKD1 εμφανίζουν βαρύτερη κλινική συμπτωματολογία και καταλήγουν νωρίτερα σε τελικού σταδίου νεφρική ανεπάρκεια.

Η ΥΠΚΝ έχει μικρότερη συχνότητα, 1:10.000-1:40.000 γεννήσεις. Οι περισσότεροι ασθενείς διαγιγνώσκονται στη νεογνική και παιδική ηλικία αλλά υπάρχουν και ήπιες περιπτώσεις της νόσου που διαγιγνώσκονται μετά την ηλικία των 20 ετών. Προκαλείται από μετάλλαξη του γονιδίου PKHD1 που εδράζεται στο χρωμόσωμα 6p21 και κωδικοποιεί την φιμπροκυστίνη. Η συχνότητα του ετερόζυγου φορέα της μετάλλαξης PKHD1 στο γενικό πληθυσμό υπολογίζεται σε 1:70.
Η παθοφυσιολογία του σχηματισμού των κύστεων δεν έχει σαφώς διευκρινισθεί. Έχουν διατυπωθεί διάφοροι μηχανισμοί όπου στηρίζονται υποσχόμενες μελλοντικές θεραπευτικές παρεμβάσεις. Οι πρωτεΐνες των αντίστοιχων γονιδίων ανιχνεύονται στο κροσσωτό επιθήλιο των νεφρικών επιθηλιακών κυττάρων και φαίνεται ότι επηρεάζεται η δομή και λειτουργικότητα των κροσσών (cilia), η έκφραση του άξονα του επιδερμικού παράγοντα αύξησης (EGFR), ο μεταβολισμός του ενδοκυττάριου Ca και cAMP καθώς και η παραγωγή της θεμελίου ουσίας. Η κλινική εικόνα στην ΕΠΚΝ, όσον αφορά και τα δύο γονίδια PKD1 και PKD2, παρουσιάζει μεγάλη ετερογένεια. Έχει διατυπωθεί η θεωρία του «διπλού χτυπήματος» που υποστηρίζει ότι η κληρονόμηση μιας μετάλλαξης στο υπεύθυνο γονίδιο είναι αναγκαία μεν αλλά όχι ικανή συνθήκη για την ανάπτυξη της πολυκυστικής παθολογίας.
Μία δεύτερη, σωματική ή επίκτητη μετάλλαξη επέρχεται και οδηγεί στην ολοκληρωτική απώλεια του δεύτερου φυσιολογικού αλληλόμορφου γονιδίου που είναι απαραίτητη και καθοριστική για να ξεκινήσει η υπερπλασία και ο πολλαπλασιασμός των κυττάρων που παραμένουν χωρίς φυσιολογική πολυκυστίνη. Η διάγνωση της EΠΚΝ στηρίζεται στην υπερηχογραφική εικόνα των νεφρών.Τυπικά ευρήματα είναι οι αυξημένων διαστάσεων υπερηχογενείς νεφροί με πολυάριθμες κύστεις διάσπαρτες σε όλο το νεφρικό παρέγχυμα αμφοτέρων των νεφρών. Το υπερηχογράφημα παραμένει η διαγνωστική μέθοδος εκλογής. Βέβαια η αξονική ή μαγνητική τομογραφία είναι πλέον ευαίσθητες αλλά σπανίως απαιτούνται. Η μοριακή διάγνωση είναι μεν δυνατή αλλά πολύπλοκη. Γίνεται είτε με ταυτοποίηση γνωστής μετάλλαξης, είτε με γενετική ανάλυση σύνδεσης και συνήθως πραγματοποιείται για την προγεννητική διάγνωση της νόσου και σε αμφίβολες περιπτώσεις δυνητικών φορέων νεφρικού μοσχεύματος.
Σημαντικά στοιχεία για τη διάγνωση της νόσου είναι η ύπαρξη ή όχι οικογενειακού ιστορικού, ο αριθμός και η μορφή των νεφρικών κύστεων και ηλικία του ασθενούς. Σε ασυμπτωματικά άτομα με θετικό οικογενειακό ιστορικό ΕΠΚΝ, εάν είναι ηλικίας <18 ετών δε συνιστάται υπερηχογραφική ανίχνευση (screening) της νόσου. Εάν είναι ενήλικες, συνιστάται υπερηχογραφικός έλεγχος εάν είναι υποψήφιοι δότες νεφρικού μοσχεύματος ή διατυπώσουν την επιθυμία να ελεγχθούν και εφ’ όσον έχει προηγηθεί ιατρική ενημέρωση. Τα υπερηχογραφικά κριτήρια που χρησιμοποιούνται για την διάγνωση της νόσου σε άτομα με θετικό οικογενειακό ιστορικό είναι: Α) μεταξύ 15 και 39 ετών, τουλάχιστον τρεις ετερόπλευρες ή αμφοτερόπλευρες κύστεις Β) μεταξύ 40 και 59 ετών τουλάχιστον δύο κύστεις σε κάθε νεφρό, Γ) σε άτομα ηλικίας > 60 ετών τουλάχιστον τέσσερις κύστεις σε κάθε νεφρό.
Επομένως προκύπτει ότι η ΕΠΚΝ, όταν υπάρχει θετικό οικογενειακό ιστορικό, δυνατόν να αποκλειστεί: α) σε άτομα ηλικίας > 40 ετών με καμία ή μόνον μία νεφρική κύστη, β) σε άτομα μεταξύ 30 και 39 ετών με ουδεμία κύστη με ψευδώς αρνητικό ποσοστό 2%, ενώ γ) σε άτομα <30 ετών, ο αποκλεισμός της νόσου με το υπερηχογράφημα νεφρών δεν είναι δυνατός.
Σε ασθενείς με αρνητικό οικογενειακό ιστορικό, δεν υπάρχει καθορισμένος αριθμός κύστεων που να επιβεβαιώνει τη διάγνωση. Η ύπαρξη πλέον των 10 κύστεων σε κάθε νεφρό, το μεγάλο μέγεθος νεφρών και η ύπαρξη ηπατικών κύστεων συνηγορούν στη διάγνωση. Περίπου 25% των ασθενών με τη συμπτωματολογία της νόσου έχουν αρνητικό οικογενειακό ιστορικό και στο 5% η νόσος οφείλεται σε νέα μετάλλαξη. Η κλινική συμπτωματολογία της νόσου διαφέρει μεταξύ των πασχόντων. Γενικά οι ασθενείς με μετάλλαξη στο PKD1 εμφανίζουν βαρύτερα συμπτώματα και σε μικρότερη ηλικία. Οι τυπικές εκδηλώσεις της νόσου είναι αρτηριακή υπέρταση, αιματουρία, λευκωματουρία, νεφρική ανεπάρκεια και άλγος στη νεφρική χώρα που δυνατόν να οφείλεται σε λιθίαση, νεφρική αιμορραγία ή ουρολοίμωξη. Επίσης δυνατόν να εμφανιστούν δευτεροπαθώς συμπτώματα από τις κύστεις στα άλλα όργανα (ήπαρ, σπλην, θυρεοειδής, επιδιδυμίδα) και ενδοκρανιακά ανευρύσματα.
Οι κυριότεροι παράγοντες που υποδηλώνουν πλέον σοβαρή εξελισσόμενη νεφρική νόσο είναι: 1) γενετικοί παράγοντες (PKD1 έναντι PKD2), 2) αρτηριακή υπέρταση, 3) εμφάνιση αιματουρίας και λευκωματουρίας σε μικρή ηλικία, 4) το άρρεν φύλο, 5) μεγάλο μέγεθος νεφρών κλπ. Οι περισσότεροι παιδιατρικοί ασθενείς είναι ασυμπτωματικοί και δυνατόν η νόσος να προβάλλει στη νεογνική περίοδο μόνο με μεγάλους υπερηχογενείς νεφρούς. Στο 17% των παιδιατρικών ασθενών αρχικά οι κύστεις είναι ετερόπλευρες, γενικά όμως όταν εμφανισθεί νεφρική συμπτωματολογία είναι παρόμοια των ενηλίκων. Οι εκδηλώσεις από εξωνεφρική εντόπιση κύστεων είναι σπάνιες στην παιδική ηλικία. Στους ασθενείς με αρνητικό οικογενειακό ιστορικό, η ηλικία του πάσχοντος, η ύπαρξη ή όχι οικογενειακού ιστορικού άλλων γενετικών διαταραχών με κυστική νεφρική προσβολή και η εμφάνιση εξωνεφρικών εκδηλώσεων βοηθούν στη διαφορική διάγνωση.
Η διαφορική διάγνωση περιλαμβάνει τις καλοήθεις απλές κύστεις, το σπογγοειδή νεφρό, την ΥΠΚΝ, την επίκτητη κυστική νόσο, την εντοπισμένη κυστική δυσπλασία, την οζώδη σκλήρυνση/νόσο von Hippel –Lindau και τη μυελώδη κυστική νόσο επικρατούντα χαρακτήρα. Πρέπει να σημειωθεί ότι εάν τεθεί η υποψία της ΕΠΚΝ σε παιδί και οι γονείς δεν παρουσιάζουν νεφρικές κύστεις στο υπερηχογράφημα νεφρών αλλά είναι ηλικίας <30 ετών, έλεγχος πρέπει να γίνει και στους γονείς των γονέων. Η προγεννητική διάγνωση στην ΕΠΚΝ είναι δυνατή με γονιδιακό έλεγχο, είτε με την ταυτοποίηση της μετάλλαξης εάν είναι γνωστή, είτε με ανάλυση σύνδεσης. Μεγάλοι υπερηχογενείς νεφροί στο έμβρυο δεν επιβεβαιώνουν την διάγνωση και βέβαια η φυσιολογική υπερηχογραφική απεικόνιση εμβρυικών νεφρών δεν αποκλείει την νόσο.
Δεν έχει τεκμηριωθεί ειδική θεραπεία για την πρόληψη ή αναστολή της εξέλιξης της νόσου. Έχουν αξιολογηθεί υποσχόμενες θεραπείες με διάφορα αποτελέσματα, που στηρίζονται στην παθοφυσιολογία του σχηματισμού των κύστεων, όπως αναστολείς των ανταγωνιστών της βαζοπρεσσίνης (tolvaptan), mTOR inhibitors (sirolimus /everolimus), σωματοστατίνη (octreotide) και μέγιστη αναστολή του άξονα ρενίνης – αλδοστερόνης (αναστολείς του μετατρεπτικού ενζύμου / ανταγωνιστές των υποδοχέων αγγειοτενσίνης).
Η αντιμετώπιση της νόσου είναι συμπτωματική και στηρίζεται σε μη ειδικά μέτρα:
1) Ο αυστηρός έλεγχος της αρτηριακής υπέρτασης προλαμβάνει την επιδείνωση της νεφρικής νόσου και μειώνει τον κίνδυνο της καρδιαγγειακής νόσου. Εάν δεν υπάρχουν αντενδείξεις, η χορήγηση ενός αναστολέα του μετατρεπτικού ενζύμου παραμένει το φάρμακο εκλογής κυρίως σε ασθενείς με λευκωματουρία. Απαιτείται όμως στενή παρακολούθηση των ασθενών υψηλού κινδύνου με μεγάλους νεφρούς και νεφρική ανεπάρκεια.
2) Η υπερλιπιδαιμία πρέπει να αντιμετωπίζεται με χορήγηση στατινών.
3) Η αυξημένη λήψη υγρών παίζει σημαντικό ρόλο γιατί αναστέλλει τα επίπεδα της βαζοπρεσσίνης που φαίνεται ότι συμμετέχει στην παθοφυσιολογία της νόσου και συνιστάται η λήψη > 3L /24ωρο.
4) Περιορισμένη λήψη πρωτεινών και άλατος.
5) Αντιμετώπιση των ουρολοιμώξεων και της φλεγμονής των κύστεων με αντιβιοτικά που διέρχονται το τοίχωμα αυτών.
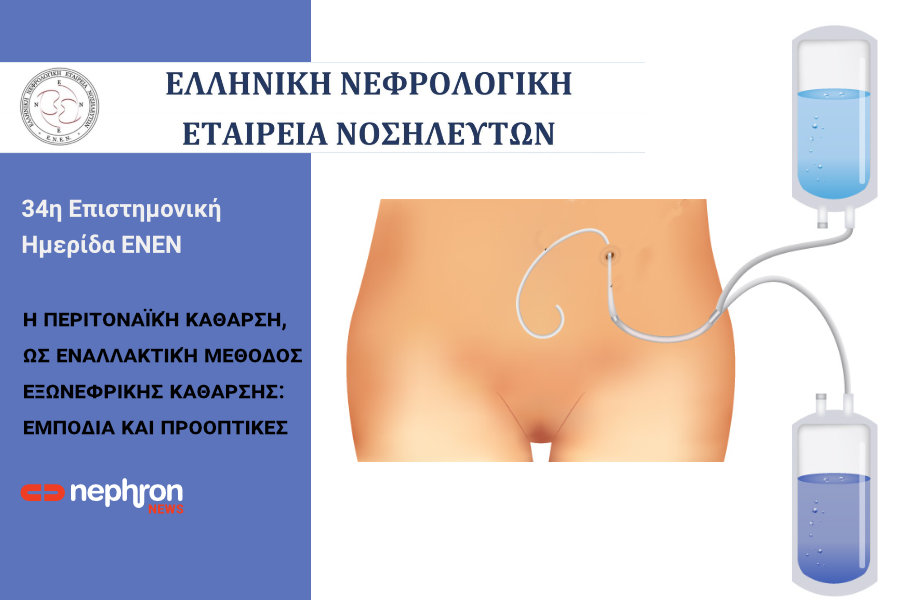
6) Αντιμετώπιση του οσφυϊκού άλγους, αιματουρίας ή ενδονεφρικής αιμορραγίας. Οι πάσχοντες από ΕΠΚΝ και ΤΣΝΑ αντιμετωπίζονται συχνότερα με αιμοκάθαρση δεδομένου ότι η εφαρμογή περιτοναικής κάθαρσης είναι προβληματική σε ασθενείς με υπερμεγέθεις κυστικούς νεφρούς. Η νεφρική μεταμόσχευση αυτών των ασθενών δεν παρουσιάζει αυξημένο ποσοστό επιπλοκών συγκριτικά με άλλους ασθενείς ΤΣΝΑ, συνιστάται όμως στην προμεταμοσχευτική αξιόλογηση, έλεγχος για την ύπαρξη ενδοκρανιακού ανευρύσματος, ποσοστό που κυμαίνεται, έως 40% των αρρώστων.
ΒΙΒΛΙΟΓΡΑΦΙΑ
- Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen. Hudson et all. N Engl J Med 2003;348:2543
- Familial hematuria. C E Kashtan. Pediatr Nephrol 2009;24 :1958
- Alport Syndrome and Thin Glomerular Basement Membrane Nephropathy. M Haas. Arch Pathol Lab Med 2009;133:224
- Clinical practice recommendations for treatment of Alport syndrome; a statement of the Alport Syndrome Research Collaborative C E Kashtan et all. Pediatr Nephrol 2013;28:5
- Thin Basement Membrane Nephropathy .K Tryggvason,J Patrakka.J Am Soc Nephrol 2006;17:813
- Genetics and Pathogenesis of Polykystic Kidney Disease. P Igarashi,S Somlo. J Am Soc Nephrol 2002;13:2384
- Autosomal dominant polycystic kidney disease:the last three years. V E Torres, P C Harris. Kidney Int 2009;76:149
- Cystic kidney disease: many ways to form cyst. H Loftus. A C M Ong. Pediatr Nephrol 2013;28:33
- The Srectrum of Polycystic Kidney Disease in Ch;ildren, K MacRae Dell. Adv Chronic Kidney Dis 2011;18:339
- Course and treatment of autosomal dominant polycystic kidney disease. A Chapman et all. http://www.uptodate.com/contents2014.
ΠΗΓΗ: Οδηγός Νεφρικής Νόσου Dialysis Living






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}